Une recherche de pointe!
Une telle recherche sur la base génétique de la SLA, à si grande échelle, n’a jamais été effectuée auparavant ! Nous voulons mener des recherches inédites sur la cause de la SLA, et, pour cela, nous avons besoin de votre aide.
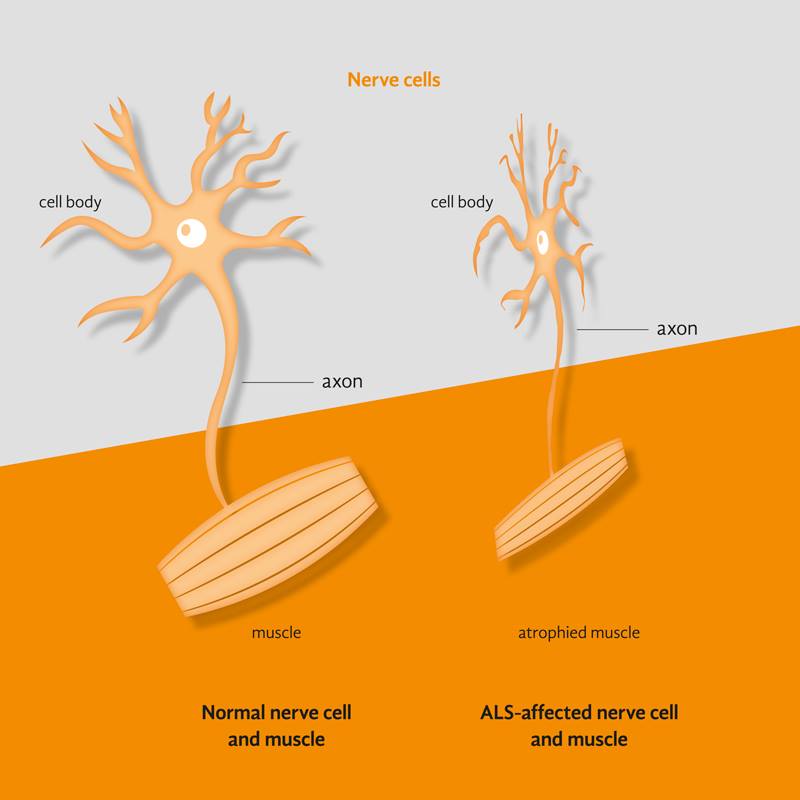
Plus de 200 000 personnes dans le monde souffrent de sclérose latérale amyotrophique, ou SLA. Il s’agit d’une maladie neuromusculaire progressive et mortelle. On ne sait que très peu de choses sur la cause de la SLA. Aucun traitement n’existe. Après le diagnostic, les patients ont une durée moyenne de survie de trois ans.
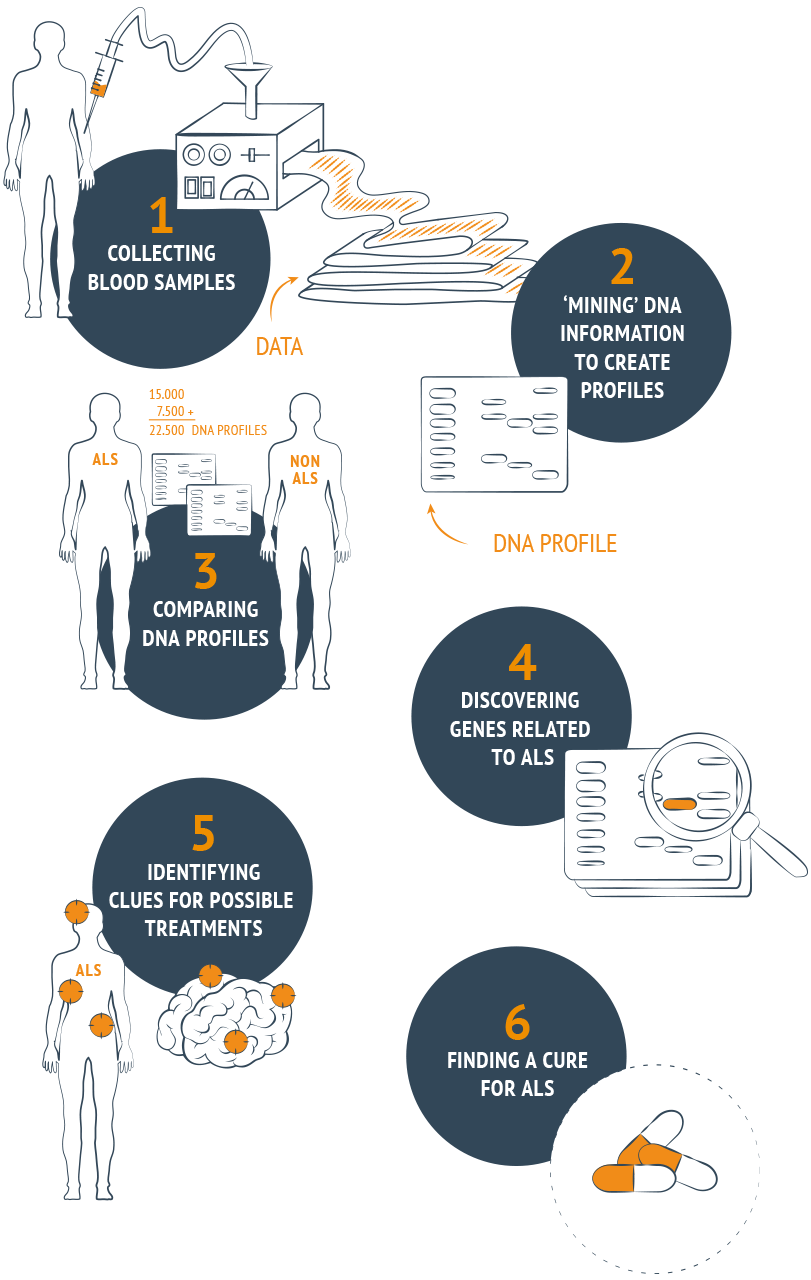
Il est presque certain que la SLA a une base génétique. ‘Project MinE’ est une étude à grande échelle sur les bases génétiques et les liens concrets vers un traitement efficace de cette maladie mortelle.
Nous voulons analyser l’ADN, déterminer les profils ADN, d’au moins 15 000 personnes atteintes de SLA et les comparer avec l’ADN de 7500 personnes de contrôle, pour identifier les différences génétiques impliquées dans l’émergence de la SLA. Il faut examiner un grand nombre de profils ADN pour récolter ces données. Ce type de recherche est très coûteux. C’est pourquoi nous avons besoin de votre soutien. Faites un don encore aujourd’hui ou commencez votre campagne. Project MinE, make it yours!